Resolution Depth Analysis for Hi-C/-like Data
Minghao Jiang
2026-01-23
Source:vignettes/depth.Rmd
depth.RmdOverview
This vignette demonstrates how to analyze Hi-C pairs data to find optimal genomic resolutions using gghic’s resolution depth functions. You’ll learn how to:
- Calculate contact depth at different bin sizes
- Assess genome coverage across resolutions
- Find optimal resolution that balances detail and completeness
- Visualize tradeoffs between resolution and coverage
When to Use These Functions
These functions are essential for:
- Pore-C & long-read data: Determining the best resolution for long DNA-DNA interactions
- Hi-C QC: Assessing data quality and sequencing depth
- Resource planning: Finding the sweet spot before expensive downstream analysis
- Publication standards: Communicating data completeness to reviewers
Getting Started
Load required libraries:
load_pkg <- function(pkgs) {
for (pkg in pkgs) suppressMessages(require(pkg, character.only = TRUE))
}
load_pkg(c("readr", "dplyr", "rappdirs", "gghic"))
#> Warning in library(package, lib.loc = lib.loc, character.only = TRUE,
#> logical.return = TRUE, : there is no package called 'readr'Example Data
We’ll create synthetic pairs data for demonstration. In practice, load from your contact matrix file:
# Create example pairs dataset
set.seed(42)
n_interactions <- 10000
pairs <- data.frame(
read_name = paste0("read_", seq_len(n_interactions)),
chrom1 = sample(c("chr1", "chr2", "chr3"), n_interactions, replace = TRUE),
pos1 = sample(1e6:10e6, n_interactions, replace = TRUE),
chrom2 = sample(c("chr1", "chr2", "chr3"), n_interactions, replace = TRUE),
pos2 = sample(1e6:10e6, n_interactions, replace = TRUE)
)
glimpse(pairs)
#> Rows: 10,000
#> Columns: 5
#> $ read_name <chr> "read_1", "read_2", "read_3", "read_4", "read_5", "read_6", …
#> $ chrom1 <chr> "chr1", "chr1", "chr1", "chr1", "chr2", "chr2", "chr2", "chr…
#> $ pos1 <int> 7501652, 9834771, 7511178, 9988046, 2376108, 8056778, 991689…
#> $ chrom2 <chr> "chr3", "chr1", "chr1", "chr2", "chr1", "chr2", "chr3", "chr…
#> $ pos2 <int> 4068533, 1789229, 6896113, 6916211, 7349759, 9964631, 150451…Expected column format:
-
read_name: Unique identifier for each interaction -
chrom1,chrom2: Chromosome names (e.g., “chr1”) -
pos1,pos2: Genomic positions in base pairs - Optional: strand, mapping quality, fragment ID (ignored by analysis functions)
Choosing Your Approach
The gghic package provides two complementary approaches for analyzing pairs data:
| Approach | Memory | Speed | When to Use |
|---|---|---|---|
In-memory
calculateResolutionDepth()
|
O(interactions) | Fastest | Files < 50% RAM, interactive analysis |
Chunked calcResDepthChunked()
|
O(unique bins) | Fast | Files > 50% RAM, automated pipelines |
In-Memory Processing (Smaller Files) - Load entire dataset into memory - Single file read (fastest) - Typical memory: ~4 bytes per interaction - Best for: Interactive exploration, visualization
Chunked Streaming (Large Files) - Read file in streaming fashion - Minimal memory footprint (~100 MB even for 100 GB files) - C-accelerated file I/O - Automatic gzip decompression - Best for: Production pipelines, server environments, very large datasets
Decision Rule: If your file size > available RAM / 2, use chunked functions.
Step 1: Calculate Resolution Depth
The calculateResolutionDepth() function bins genomic
positions and counts unique interactions per bin:
# Calculate depth at 10kb resolution
depth_10kb <- calculateResolutionDepth(pairs, bin_size = 10000)
depth_10kb
#> # A tibble: 2,699 × 3
#> chrom bin count
#> <chr> <int> <int>
#> 1 chr1 845 12
#> 2 chr2 812 6
#> 3 chr3 779 7
#> 4 chr1 846 7
#> 5 chr2 813 6
#> 6 chr3 780 4
#> 7 chr1 847 7
#> 8 chr3 781 8
#> 9 chr2 814 6
#> 10 chr1 848 9
#> # ℹ 2,689 more rows
# Summary statistics
summary(depth_10kb$count)
#> Min. 1st Qu. Median Mean 3rd Qu. Max.
#> 1.00 5.00 7.00 7.41 9.00 19.00What this returns:
-
chrom: Chromosome name -
bin: Bin number (position / bin_size, rounded up) -
count: Number of unique interactions in this bin
Interpreting the results:
- High counts = well-sequenced regions
- Low counts = sparse or unmappable regions
- Zero-count bins = gaps (excluded from output)
Step 2: Assess Genome Coverage
The calculateGenomeCoverage() function determines what
fraction of bins meet a minimum contact threshold:
# Calculate coverage at multiple resolutions
resolutions <- c(5e3, 10e3, 25e3, 50e3, 100e3, 500e3)
coverage_df <- data.frame(
resolution_kb = resolutions / 1e3,
n_bins = NA,
coverage = NA
)
for (i in seq_along(resolutions)) {
depth <- calculateResolutionDepth(pairs, resolutions[i])
coverage_df$n_bins[i] <- nrow(depth)
coverage_df$coverage[i] <- calculateGenomeCoverage(
pairs, resolutions[i],
min_contacts = 50
)
}
coverage_df$coverage_pct <- sprintf("%.1f%%", coverage_df$coverage * 100)
coverage_df
#> resolution_kb n_bins coverage coverage_pct
#> 1 5 5273 0.00000000 0.0%
#> 2 10 2699 0.00000000 0.0%
#> 3 25 1080 0.00000000 0.0%
#> 4 50 540 0.02777778 2.8%
#> 5 100 270 0.99629630 99.6%
#> 6 500 54 1.00000000 100.0%Interpreting coverage:
- High coverage (>80%) at small bins: Well-sequenced, good resolution potential
- Low coverage (<50%) at large bins: Sparse data, may need deeper sequencing
- Coverage curve shape: Shows sequencing depth and data quality
Step 3: Find Optimal Resolution
Use binary search to find the smallest bin size achieving your target coverage:
# Find resolution with ~80% coverage
optimal_bin <- findOptimalResolution(
pairs,
min_bin = 1e3, # Start at 1 Kb
max_bin = 1e6, # Search up to 1 Mb
target_coverage = 0.8,
min_contacts = 50
)
#> Iteration 1: Testing bin size 500,500 bp: 98.18% coverage
#> Iteration 2: Testing bin size 250,749 bp: 97.30% coverage
#> Iteration 3: Testing bin size 125,874 bp: 97.72% coverage
#> Iteration 4: Testing bin size 63,436 bp: 36.60% coverage
#> Iteration 5: Testing bin size 94,655 bp: 97.92% coverage
#> Iteration 6: Testing bin size 79,045 bp: 88.41% coverage
#> Iteration 7: Testing bin size 71,240 bp: 64.83% coverage
#> Iteration 8: Testing bin size 75,142 bp: 77.13% coverage
#> Iteration 9: Testing bin size 77,093 bp: 83.85% coverage
#> Iteration 10: Testing bin size 76,117 bp: 81.23% coverage
#> Iteration 11: Testing bin size 75,629 bp: 78.61% coverage
#> Iteration 12: Testing bin size 75,873 bp: 81.23% coverage
#> Iteration 13: Testing bin size 75,751 bp: 79.55% coverage
#> Iteration 14: Testing bin size 75,812 bp: 79.27% coverage
#> Iteration 15: Testing bin size 75,842 bp: 80.67% coverage
#> Iteration 16: Testing bin size 75,827 bp: 80.39% coverage
#> Iteration 17: Testing bin size 75,819 bp: 79.83% coverage
#> Iteration 18: Testing bin size 75,823 bp: 80.11% coverage
#> Iteration 19: Testing bin size 75,821 bp: 79.55% coverage
#> Iteration 20: Testing bin size 75,822 bp: 79.55% coverage
cat(sprintf(
"Optimal bin size: %d bp (%.1f Kb)\n", optimal_bin, optimal_bin / 1000
))
#> Optimal bin size: 75823 bp (75.8 Kb)
# Verify the result
actual_coverage <- calculateGenomeCoverage(
pairs, optimal_bin,
min_contacts = 50
)
cat(sprintf("Actual coverage: %.2f%%\n", actual_coverage * 100))
#> Actual coverage: 80.11%How it works:
- Binary search across the bin size range
- Tests each candidate bin size
- Returns smallest bin that meets target coverage
- Typically completes in 10-15 iterations
Pro tip: Start with
target_coverage = 0.8 (80%). Adjust based on your analysis
needs.
Step 4: Visualize Coverage Distribution
Create a boxplot showing how contact counts vary across resolutions:
plotResolutionCoverage(
pairs,
bin_sizes = c(5e3, 10e3, 25e3, 50e3, 100e3, 500e3),
min_contacts = 50,
title = "Contact Distribution by Resolution"
)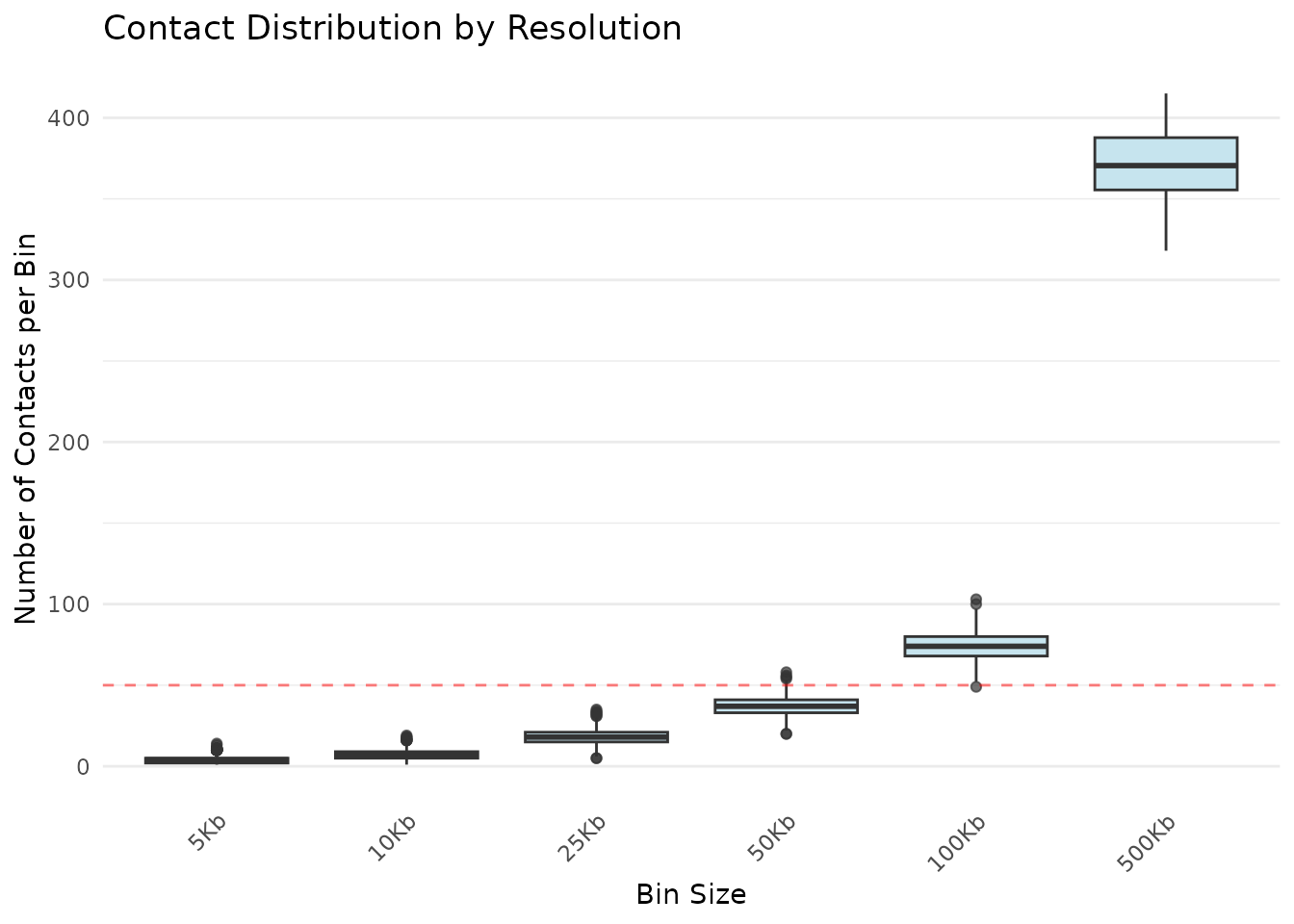
Distribution of contacts per bin at different resolutions
What to look for:
- Widening boxes: Decreasing contacts with larger bins (expected)
- Outliers (dots): Unusually enriched regions (e.g., centromeres, pericentromeres)
- Median position: Typical coverage at that resolution
- Whisker length: Range of contact values (high whiskers = variable coverage)
Step 5: Plot Coverage vs Resolution
Create a line plot showing how coverage changes with resolution:
plotCoverageCurve(
pairs,
bin_sizes = c(5e3, 10e3, 25e3, 50e3, 100e3, 500e3, 1e6),
min_contacts = 50,
title = "Coverage vs Resolution"
)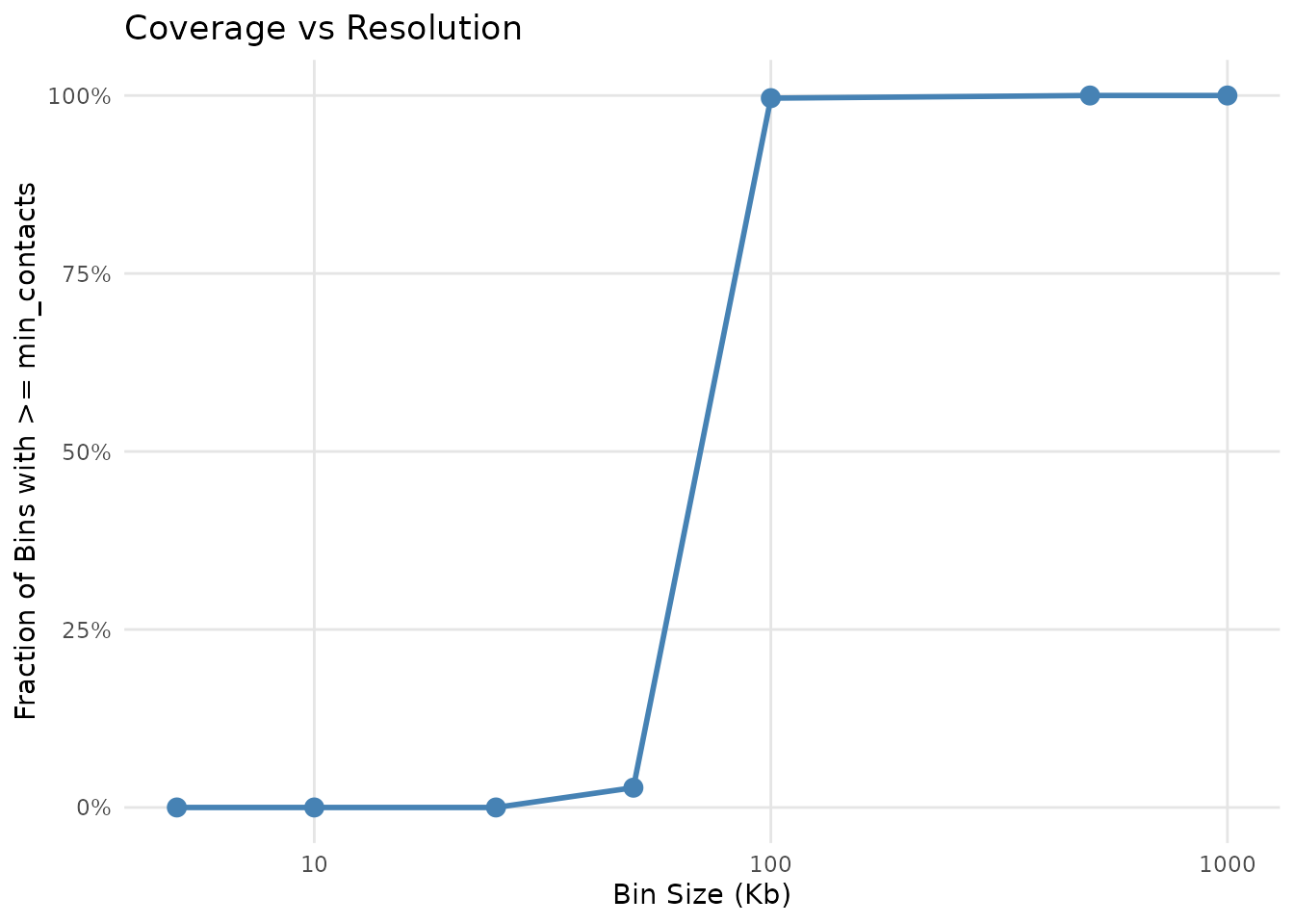
Genome coverage as a function of bin size
Curve interpretation:
- Sharp drop at small bins: Limited sequencing depth
- Plateau region: Where additional bin size doesn’t improve coverage much
- “Knee” of curve: The optimal resolution sweet spot
- Steep regions: Good resolution potential at that bin size
Processing Large Files (100 GB+)
⚠️ Note: The code examples in this section are provided for reference but are not executed in this vignette.
For files that don’t fit in R memory, use the _chunked
variants which stream the file with C-level I/O:
# Download example file with caching
cache_dir <- rappdirs::user_cache_dir("gghic")
dir.create(cache_dir, recursive = TRUE, showWarnings = FALSE)
pairs_file <- file.path(cache_dir, "test.txt.gz")
download_url <- "https://www.dropbox.com/scl/fi/yc4axg1mf2i9zylg3d0oe/test.txt.gz?rlkey=sdsdhsnixo01koo38d242y4c8&st=t4rn0js0&dl=1"
if (!file.exists(pairs_file)) {
message("Downloading test data to cache directory...")
download.file(
download_url, pairs_file,
method = "auto", quiet = TRUE
)
message("Downloaded to: ", pairs_file)
} else {
message("Using cached file: ", pairs_file)
}Approach 1: Direct File Reading (Simple but Slower)
Read the file each time a function is called:
# Each function reads the file independently
opt_bin <- findOptResChunked(
pairs_file = pairs_file,
min_bin = 1e3,
max_bin = 5e4,
target_coverage = 0.8,
min_contacts = 1000
)
# Reads file again
coverage <- calcGenomeCovChunked(
pairs_file = pairs_file,
bin_size = opt_bin,
min_contacts = 1000
)
# Reads file again
depth <- calcResDepthChunked(
pairs_file = pairs_file,
bin_size = opt_bin
)Problem: For a 10 GB file, you’re reading 10 GB three times (30 GB total I/O)!
Approach 2: Cache Once, Reuse Many Times (FASTEST!)
Read the file once into C memory, then reuse the cached data:
# 1. READ FILE INTO C MEMORY ONCE ----
# This takes time initially but creates a persistent cache
cache <- readPairsCache(pairs_file)
cat("File loaded into C memory cache\n")
# 2. FIND OPTIMAL RESOLUTION (using cache) ----
# This is now instant - no file I/O!
opt_bin <- findOptResChunked(
cache = cache,
min_bin = 1e3,
max_bin = 5e4,
target_coverage = 0.8,
min_contacts = 1000
)
cat("Optimal resolution:", opt_bin / 1e3, "Kb\n")
# 3. CHECK COVERAGE (using cache) ----
# Also instant!
coverage <- calcGenomeCovChunked(
cache = cache,
bin_size = opt_bin,
min_contacts = 1000
)
cat("Coverage:", sprintf("%.1f%%\n", coverage * 100))
# 4. EXTRACT BINNED DEPTHS (using cache) ----
# Still instant!
depth <- calcResDepthChunked(
cache = cache,
bin_size = opt_bin
)
# 5. TRY DIFFERENT BIN SIZES (no additional I/O!) ----
depth_5kb <- calcResDepthChunked(cache = cache, bin_size = 5000)
depth_10kb <- calcResDepthChunked(cache = cache, bin_size = 10000)
depth_50kb <- calcResDepthChunked(cache = cache, bin_size = 50000)
# Cache is automatically freed when removed or R session ends
rm(cache)
gc()Performance: For a 10 GB file: - Without cache: ~30 seconds × 3 operations = 90 seconds - With cache: ~30 seconds (once) + ~0.1 seconds × operations = 30 seconds
3x faster! And the speedup increases with more operations.
Approach 3: Get Cache from findOptResChunked
Let findOptResChunked() return the cache for subsequent
use:
# Find optimal resolution and get cache back
result <- findOptResChunked(
pairs_file = pairs_file,
min_bin = 1e3,
max_bin = 5e4,
target_coverage = 0.8,
min_contacts = 1000,
return_cache = TRUE # Get the cache back!
)
# Extract results
opt_bin <- result$bin_size
cache <- result$cache
cat("Optimal resolution:", opt_bin / 1e3, "Kb\n")
# Now reuse the cache for other operations
coverage <- calcGenomeCovChunked(
cache = cache,
bin_size = opt_bin,
min_contacts = 1000
)
depth <- calcResDepthChunked(
cache = cache,
bin_size = opt_bin
)When to Use Each Approach
| Approach | Best For | I/O Operations |
|---|---|---|
| Direct File | Single analysis, simple scripts | N operations |
| Explicit Cache | Interactive exploration, multiple analyses | 1 operation |
| Return Cache | When starting with findOptResChunked()
|
1 operation |
Rule of thumb: If you’ll call more than one chunked function, use the cache!
Interpreting Your Results
What the Coverage Curve Tells You
Sharp drop at small bins - Data is sparse at high resolution - Limited sequencing depth or short reads - Consider deeper sequencing or larger bins
Plateau region - Coverage maxes out (can’t improve by increasing bin size) - Indicates natural sequencing depth ceiling - Optimal resolution likely at transition point
High coverage at large bins (>80%) - Well-sequenced dataset - Can use smaller bins safely - Good candidate for publication-quality analysis
Low coverage at large bins (<50%) - Sparse data - May need additional sequencing - Consider quality filtering
Next Steps
Once you’ve found your optimal resolution, explore other gghic functions for visualization:
-
geom_hic(): Create interaction scatter plots -
geom_loop(): Highlight chromatin loops -
geom_tad(): Show topologically associating domains -
geom_ideogram(): Add chromosome context -
theme_hic(): Specialized theme for Hi-C plots
See the main gghic vignette for complete examples.
FAQ
Q: Why does coverage decrease with larger bins? A: By definition—larger bins aggregate more positions but contact counts don’t increase proportionally. Coverage measures the fraction of bins with sufficient contacts.
Q: Should I use chunked functions for small files? A: No, use in-memory functions—they’re simpler and faster for files that fit in RAM.
Q: Can I use these functions on non-Hi-C data? A: Yes, any “pairs” data with (chrom, position) coordinates works: Pore-C, 4C, 5C, etc.
Session Info
sessionInfo()
#> R version 4.5.2 (2025-10-31)
#> Platform: x86_64-pc-linux-gnu
#> Running under: Ubuntu 24.04.3 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
#> [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
#> [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
#> [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] gghic_0.2.1 rappdirs_0.3.4 dplyr_1.1.4
#>
#> loaded via a namespace (and not attached):
#> [1] SummarizedExperiment_1.40.0 gtable_0.3.6
#> [3] rjson_0.2.23 xfun_0.56
#> [5] bslib_0.9.0 ggplot2_4.0.1
#> [7] htmlwidgets_1.6.4 rhdf5_2.54.1
#> [9] Biobase_2.70.0 lattice_0.22-7
#> [11] bitops_1.0-9 rhdf5filters_1.22.0
#> [13] vctrs_0.7.0 tools_4.5.2
#> [15] generics_0.1.4 parallel_4.5.2
#> [17] stats4_4.5.2 curl_7.0.0
#> [19] tibble_3.3.1 pkgconfig_2.0.3
#> [21] Matrix_1.7-4 RColorBrewer_1.1-3
#> [23] cigarillo_1.0.0 S7_0.2.1
#> [25] desc_1.4.3 S4Vectors_0.48.0
#> [27] lifecycle_1.0.5 compiler_4.5.2
#> [29] farver_2.1.2 Rsamtools_2.26.0
#> [31] Biostrings_2.78.0 textshaping_1.0.4
#> [33] codetools_0.2-20 Seqinfo_1.0.0
#> [35] InteractionSet_1.38.0 htmltools_0.5.9
#> [37] sass_0.4.10 RCurl_1.98-1.17
#> [39] yaml_2.3.12 tidyr_1.3.2
#> [41] crayon_1.5.3 pillar_1.11.1
#> [43] pkgdown_2.2.0 jquerylib_0.1.4
#> [45] BiocParallel_1.44.0 DelayedArray_0.36.0
#> [47] cachem_1.1.0 abind_1.4-8
#> [49] tidyselect_1.2.1 digest_0.6.39
#> [51] purrr_1.2.1 restfulr_0.0.16
#> [53] labeling_0.4.3 fastmap_1.2.0
#> [55] grid_4.5.2 cli_3.6.5
#> [57] SparseArray_1.10.8 magrittr_2.0.4
#> [59] S4Arrays_1.10.1 utf8_1.2.6
#> [61] dichromat_2.0-0.1 XML_3.99-0.20
#> [63] withr_3.0.2 scales_1.4.0
#> [65] rmarkdown_2.30 XVector_0.50.0
#> [67] httr_1.4.7 matrixStats_1.5.0
#> [69] ragg_1.5.0 evaluate_1.0.5
#> [71] knitr_1.51 BiocIO_1.20.0
#> [73] GenomicRanges_1.62.1 IRanges_2.44.0
#> [75] rtracklayer_1.70.1 rlang_1.1.7
#> [77] Rcpp_1.1.1 glue_1.8.0
#> [79] BiocGenerics_0.56.0 jsonlite_2.0.0
#> [81] R6_2.6.1 Rhdf5lib_1.32.0
#> [83] GenomicAlignments_1.46.0 MatrixGenerics_1.22.0
#> [85] systemfonts_1.3.1 fs_1.6.6